Phenotypic Mutation 'dee_dee' (pdf version)
| Allele | dee_dee |
|---|
| Mutation Type |
nonsense
|
|---|
| Chromosome | 10 |
|---|
| Coordinate | 60,143,835 bp (GRCm39) |
|---|
| Base Change | G ⇒ T (forward strand) |
|---|
| Gene |
Cdh23
|
| Gene Name | cadherin related 23 (otocadherin) |
|---|
| Synonym(s) | bob, sals, USH1D, ahl, mdfw, nmf252, 4930542A03Rik, nmf112, nmf181 |
|---|
| Chromosomal Location |
60,138,527-60,532,269 bp (-) (GRCm39)
|
|---|
| MGI Phenotype |
FUNCTION: [Summary is not available for the mouse gene. This summary is for the human ortholog.] This gene is a member of the cadherin superfamily, whose genes encode calcium dependent cell-cell adhesion glycoproteins. The encoded protein is thought to be involved in stereocilia organization and hair bundle formation. The gene is located in a region containing the human deafness loci DFNB12 and USH1D. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of this cadherin-like gene. Upregulation of this gene may also be associated with breast cancer. Alternative splice variants encoding different isoforms have been described. [provided by RefSeq, May 2013]
PHENOTYPE: Mutant mice exhibit circling behavior, tilting of the head and are deaf. Mice homozygous for a targeted knock-out exhibit abnormal outer hair cells morphology. [provided by MGI curators]
|
| Accession Number | NCBI RefSeq: NM_023370.3; NM_001252635.1; ENSMUST00000105461; MGI:1890219
|
|---|
| Mapped | Yes |
|---|
| Amino Acid Change |
Tyrosine changed to Stop codon
|
|---|
| Institutional Source | Beutler Lab |
|---|
| Gene Model |
predicted gene model for protein(s):
[ENSMUSP00000072973]
[ENSMUSP00000101101]
[ENSMUSP00000101102]
[ENSMUSP00000101103]
[ENSMUSP00000101104]
|
|---|
| AlphaFold |
no structure available at present |
| SMART Domains |
Protein: ENSMUSP00000072973
Gene: ENSMUSG00000012819
AA Change: Y2922*
| Domain | Start | End | E-Value | Type |
|---|
|
signal peptide
|
1 |
23 |
N/A |
INTRINSIC |
|
CA
|
55 |
130 |
5.15e-13 |
SMART |
|
CA
|
154 |
234 |
3.19e-18 |
SMART |
|
CA
|
258 |
346 |
2.03e-11 |
SMART |
|
CA
|
371 |
458 |
8.11e-11 |
SMART |
|
CA
|
482 |
559 |
1.04e-22 |
SMART |
|
CA
|
583 |
669 |
3.55e-25 |
SMART |
|
CA
|
693 |
776 |
2.04e-25 |
SMART |
|
CA
|
800 |
888 |
5.03e-16 |
SMART |
|
CA
|
912 |
993 |
1.05e-27 |
SMART |
|
CA
|
1017 |
1100 |
1.99e-19 |
SMART |
|
CA
|
1124 |
1206 |
6.94e-19 |
SMART |
|
CA
|
1231 |
1311 |
1.99e-19 |
SMART |
|
CA
|
1335 |
1415 |
1.21e-18 |
SMART |
|
CA
|
1440 |
1524 |
2.38e-26 |
SMART |
|
CA
|
1549 |
1631 |
6.27e-26 |
SMART |
|
CA
|
1656 |
1741 |
6.99e-24 |
SMART |
|
CA
|
1765 |
1848 |
3.49e-24 |
SMART |
|
CA
|
1872 |
1956 |
2.78e-18 |
SMART |
|
CA
|
1984 |
2066 |
5.6e-14 |
SMART |
|
CA
|
2090 |
2171 |
2.59e-27 |
SMART |
|
CA
|
2195 |
2290 |
2.87e-11 |
SMART |
|
CA
|
2317 |
2399 |
1.01e-20 |
SMART |
|
CA
|
2423 |
2506 |
1.09e-25 |
SMART |
|
CA
|
2530 |
2608 |
7.91e-23 |
SMART |
|
CA
|
2634 |
2719 |
1.06e-23 |
SMART |
|
CA
|
2750 |
2843 |
2e-10 |
SMART |
|
Blast:CA
|
2867 |
2956 |
4e-51 |
BLAST |
|
transmembrane domain
|
3067 |
3089 |
N/A |
INTRINSIC |
|
|---|
| Predicted Effect |
probably null
|
|---|
| SMART Domains |
Protein: ENSMUSP00000101101
Gene: ENSMUSG00000012819
AA Change: Y2923*
| Domain | Start | End | E-Value | Type |
|---|
|
signal peptide
|
1 |
23 |
N/A |
INTRINSIC |
|
CA
|
55 |
130 |
5.15e-13 |
SMART |
|
CA
|
154 |
234 |
3.19e-18 |
SMART |
|
CA
|
258 |
346 |
2.03e-11 |
SMART |
|
CA
|
371 |
458 |
1.25e-11 |
SMART |
|
CA
|
482 |
559 |
1.04e-22 |
SMART |
|
CA
|
583 |
669 |
3.55e-25 |
SMART |
|
CA
|
693 |
776 |
2.04e-25 |
SMART |
|
CA
|
800 |
888 |
5.03e-16 |
SMART |
|
CA
|
912 |
993 |
1.05e-27 |
SMART |
|
CA
|
1017 |
1100 |
1.99e-19 |
SMART |
|
CA
|
1124 |
1206 |
6.94e-19 |
SMART |
|
CA
|
1231 |
1311 |
1.99e-19 |
SMART |
|
CA
|
1335 |
1416 |
5.26e-19 |
SMART |
|
CA
|
1441 |
1525 |
2.38e-26 |
SMART |
|
CA
|
1550 |
1632 |
6.27e-26 |
SMART |
|
CA
|
1657 |
1742 |
6.99e-24 |
SMART |
|
CA
|
1766 |
1849 |
3.49e-24 |
SMART |
|
CA
|
1873 |
1957 |
2.78e-18 |
SMART |
|
CA
|
1985 |
2067 |
5.6e-14 |
SMART |
|
CA
|
2091 |
2172 |
2.59e-27 |
SMART |
|
CA
|
2196 |
2291 |
2.87e-11 |
SMART |
|
CA
|
2318 |
2400 |
1.01e-20 |
SMART |
|
CA
|
2424 |
2507 |
1.09e-25 |
SMART |
|
CA
|
2531 |
2609 |
7.91e-23 |
SMART |
|
CA
|
2635 |
2720 |
1.06e-23 |
SMART |
|
CA
|
2751 |
2844 |
2e-10 |
SMART |
|
Blast:CA
|
2868 |
2957 |
4e-51 |
BLAST |
|
transmembrane domain
|
3068 |
3090 |
N/A |
INTRINSIC |
|
|---|
| Predicted Effect |
probably null
|
|---|
| SMART Domains |
Protein: ENSMUSP00000101102
Gene: ENSMUSG00000012819
AA Change: Y2925*
| Domain | Start | End | E-Value | Type |
|---|
|
signal peptide
|
1 |
23 |
N/A |
INTRINSIC |
|
CA
|
55 |
130 |
5.15e-13 |
SMART |
|
CA
|
154 |
234 |
3.19e-18 |
SMART |
|
CA
|
261 |
349 |
2.03e-11 |
SMART |
|
CA
|
374 |
461 |
8.11e-11 |
SMART |
|
CA
|
485 |
562 |
1.04e-22 |
SMART |
|
CA
|
586 |
672 |
3.55e-25 |
SMART |
|
CA
|
696 |
779 |
2.04e-25 |
SMART |
|
CA
|
803 |
891 |
5.03e-16 |
SMART |
|
CA
|
915 |
996 |
1.05e-27 |
SMART |
|
CA
|
1020 |
1103 |
1.99e-19 |
SMART |
|
CA
|
1127 |
1209 |
6.94e-19 |
SMART |
|
CA
|
1234 |
1314 |
1.99e-19 |
SMART |
|
CA
|
1338 |
1418 |
1.21e-18 |
SMART |
|
CA
|
1443 |
1527 |
2.38e-26 |
SMART |
|
CA
|
1552 |
1634 |
6.27e-26 |
SMART |
|
CA
|
1659 |
1744 |
6.99e-24 |
SMART |
|
CA
|
1768 |
1851 |
3.49e-24 |
SMART |
|
CA
|
1875 |
1959 |
2.78e-18 |
SMART |
|
CA
|
1987 |
2069 |
5.6e-14 |
SMART |
|
CA
|
2093 |
2174 |
2.59e-27 |
SMART |
|
CA
|
2198 |
2293 |
2.87e-11 |
SMART |
|
CA
|
2320 |
2402 |
1.01e-20 |
SMART |
|
CA
|
2426 |
2509 |
1.09e-25 |
SMART |
|
CA
|
2533 |
2611 |
7.91e-23 |
SMART |
|
CA
|
2637 |
2722 |
1.06e-23 |
SMART |
|
CA
|
2753 |
2846 |
2e-10 |
SMART |
|
Blast:CA
|
2870 |
2959 |
4e-51 |
BLAST |
|
transmembrane domain
|
3070 |
3092 |
N/A |
INTRINSIC |
|
|---|
| Predicted Effect |
probably null
|
|---|
| SMART Domains |
Protein: ENSMUSP00000101103
Gene: ENSMUSG00000012819
AA Change: Y2923*
| Domain | Start | End | E-Value | Type |
|---|
|
signal peptide
|
1 |
23 |
N/A |
INTRINSIC |
|
CA
|
55 |
130 |
5.15e-13 |
SMART |
|
CA
|
154 |
234 |
3.19e-18 |
SMART |
|
CA
|
258 |
346 |
2.03e-11 |
SMART |
|
CA
|
371 |
458 |
1.25e-11 |
SMART |
|
CA
|
482 |
559 |
1.04e-22 |
SMART |
|
CA
|
583 |
669 |
3.55e-25 |
SMART |
|
CA
|
693 |
776 |
2.04e-25 |
SMART |
|
CA
|
800 |
888 |
5.03e-16 |
SMART |
|
CA
|
912 |
993 |
1.05e-27 |
SMART |
|
CA
|
1017 |
1100 |
1.99e-19 |
SMART |
|
CA
|
1124 |
1206 |
6.94e-19 |
SMART |
|
CA
|
1231 |
1311 |
1.99e-19 |
SMART |
|
CA
|
1335 |
1416 |
5.26e-19 |
SMART |
|
CA
|
1441 |
1525 |
2.38e-26 |
SMART |
|
CA
|
1550 |
1632 |
6.27e-26 |
SMART |
|
CA
|
1657 |
1742 |
6.99e-24 |
SMART |
|
CA
|
1766 |
1849 |
3.49e-24 |
SMART |
|
CA
|
1873 |
1957 |
2.78e-18 |
SMART |
|
CA
|
1985 |
2067 |
5.6e-14 |
SMART |
|
CA
|
2091 |
2172 |
2.59e-27 |
SMART |
|
CA
|
2196 |
2291 |
2.87e-11 |
SMART |
|
CA
|
2318 |
2400 |
1.01e-20 |
SMART |
|
CA
|
2424 |
2507 |
1.09e-25 |
SMART |
|
CA
|
2531 |
2609 |
7.91e-23 |
SMART |
|
CA
|
2635 |
2720 |
1.06e-23 |
SMART |
|
CA
|
2751 |
2844 |
2e-10 |
SMART |
|
Blast:CA
|
2868 |
2957 |
4e-51 |
BLAST |
|
transmembrane domain
|
3068 |
3090 |
N/A |
INTRINSIC |
|
|---|
| Predicted Effect |
probably null
|
|---|
| SMART Domains |
Protein: ENSMUSP00000101104
Gene: ENSMUSG00000012819
AA Change: Y2921*
| Domain | Start | End | E-Value | Type |
|---|
|
signal peptide
|
1 |
23 |
N/A |
INTRINSIC |
|
CA
|
55 |
130 |
5.15e-13 |
SMART |
|
CA
|
154 |
234 |
3.19e-18 |
SMART |
|
CA
|
258 |
346 |
2.03e-11 |
SMART |
|
CA
|
371 |
456 |
3.58e-12 |
SMART |
|
CA
|
480 |
557 |
1.04e-22 |
SMART |
|
CA
|
581 |
667 |
3.55e-25 |
SMART |
|
CA
|
691 |
774 |
2.04e-25 |
SMART |
|
CA
|
798 |
886 |
5.03e-16 |
SMART |
|
CA
|
910 |
991 |
1.05e-27 |
SMART |
|
CA
|
1015 |
1098 |
1.99e-19 |
SMART |
|
CA
|
1122 |
1204 |
6.94e-19 |
SMART |
|
CA
|
1229 |
1309 |
1.99e-19 |
SMART |
|
CA
|
1333 |
1414 |
5.26e-19 |
SMART |
|
CA
|
1439 |
1523 |
2.38e-26 |
SMART |
|
CA
|
1548 |
1630 |
6.27e-26 |
SMART |
|
CA
|
1655 |
1740 |
6.99e-24 |
SMART |
|
CA
|
1764 |
1847 |
3.49e-24 |
SMART |
|
CA
|
1871 |
1955 |
2.78e-18 |
SMART |
|
CA
|
1983 |
2065 |
5.6e-14 |
SMART |
|
CA
|
2089 |
2170 |
2.59e-27 |
SMART |
|
CA
|
2194 |
2289 |
2.87e-11 |
SMART |
|
CA
|
2316 |
2398 |
1.01e-20 |
SMART |
|
CA
|
2422 |
2505 |
1.09e-25 |
SMART |
|
CA
|
2529 |
2607 |
7.91e-23 |
SMART |
|
CA
|
2633 |
2718 |
1.06e-23 |
SMART |
|
CA
|
2749 |
2842 |
2e-10 |
SMART |
|
Blast:CA
|
2866 |
2955 |
3e-51 |
BLAST |
|
transmembrane domain
|
3066 |
3088 |
N/A |
INTRINSIC |
|
|---|
| Predicted Effect |
probably null
|
|---|
| Meta Mutation Damage Score |
0.9700  |
|---|
| Is this an essential gene? |
Possibly nonessential (E-score: 0.428) |
|---|
| Phenotypic Category |
Autosomal Recessive |
| Candidate Explorer Status |
loading ... |
Single pedigree
Linkage Analysis Data
|
|
| Penetrance | |
|---|
| Alleles Listed at MGI | All alleles(29) : Targeted(5) Gene trapped(2) Spontaneous(12) Chemically induced(7) QTL(3)
|
|---|
| Lab Alleles |
| Allele | Source | Chr | Coord | Type | Predicted Effect | PPH Score |
|---|
|
IGL00229:Cdh23
|
APN |
10 |
60359327 |
missense |
probably benign |
0.03 |
|
IGL00429:Cdh23
|
APN |
10 |
60256920 |
missense |
probably damaging |
0.97 |
|
IGL01014:Cdh23
|
APN |
10 |
60143301 |
missense |
probably damaging |
0.99 |
|
IGL01284:Cdh23
|
APN |
10 |
60301876 |
missense |
possibly damaging |
0.95 |
|
IGL01305:Cdh23
|
APN |
10 |
60148403 |
missense |
probably damaging |
1.00 |
|
IGL01367:Cdh23
|
APN |
10 |
60146566 |
missense |
probably damaging |
1.00 |
|
IGL01396:Cdh23
|
APN |
10 |
60220848 |
missense |
possibly damaging |
0.93 |
|
IGL01412:Cdh23
|
APN |
10 |
60150473 |
missense |
probably damaging |
1.00 |
|
IGL01461:Cdh23
|
APN |
10 |
60244926 |
missense |
possibly damaging |
0.53 |
|
IGL01469:Cdh23
|
APN |
10 |
60433504 |
missense |
probably benign |
0.03 |
|
IGL01695:Cdh23
|
APN |
10 |
60167612 |
missense |
probably benign |
0.20 |
|
IGL01734:Cdh23
|
APN |
10 |
60139292 |
missense |
probably benign |
|
|
IGL01767:Cdh23
|
APN |
10 |
60151503 |
missense |
probably damaging |
1.00 |
|
IGL01796:Cdh23
|
APN |
10 |
60146916 |
missense |
probably benign |
0.31 |
|
IGL01843:Cdh23
|
APN |
10 |
60255598 |
splice site |
probably null |
|
|
IGL02025:Cdh23
|
APN |
10 |
60220922 |
missense |
probably damaging |
1.00 |
|
IGL02071:Cdh23
|
APN |
10 |
60359339 |
missense |
possibly damaging |
0.93 |
|
IGL02160:Cdh23
|
APN |
10 |
60433544 |
splice site |
probably benign |
|
|
IGL02175:Cdh23
|
APN |
10 |
60167087 |
missense |
possibly damaging |
0.92 |
|
IGL02220:Cdh23
|
APN |
10 |
60140903 |
missense |
probably damaging |
1.00 |
|
IGL02302:Cdh23
|
APN |
10 |
60159302 |
missense |
possibly damaging |
0.87 |
|
IGL02331:Cdh23
|
APN |
10 |
60301322 |
missense |
probably damaging |
0.99 |
|
IGL02452:Cdh23
|
APN |
10 |
60153721 |
missense |
probably damaging |
0.99 |
|
IGL02499:Cdh23
|
APN |
10 |
60220958 |
missense |
probably damaging |
1.00 |
|
IGL02548:Cdh23
|
APN |
10 |
60485901 |
missense |
probably benign |
0.37 |
|
IGL02593:Cdh23
|
APN |
10 |
60301774 |
splice site |
probably benign |
|
|
IGL02626:Cdh23
|
APN |
10 |
60227580 |
missense |
probably damaging |
1.00 |
|
IGL02951:Cdh23
|
APN |
10 |
60147143 |
missense |
probably damaging |
1.00 |
|
IGL03145:Cdh23
|
APN |
10 |
60212593 |
missense |
probably damaging |
0.99 |
|
hersey
|
UTSW |
10 |
60143815 |
missense |
probably damaging |
1.00 |
|
ANU22:Cdh23
|
UTSW |
10 |
60148403 |
missense |
probably damaging |
1.00 |
|
IGL02980:Cdh23
|
UTSW |
10 |
60150399 |
missense |
probably damaging |
1.00 |
|
PIT4362001:Cdh23
|
UTSW |
10 |
60301237 |
missense |
probably benign |
0.15 |
|
R0013:Cdh23
|
UTSW |
10 |
60248952 |
missense |
possibly damaging |
0.90 |
|
R0045:Cdh23
|
UTSW |
10 |
60366757 |
missense |
probably damaging |
1.00 |
|
R0045:Cdh23
|
UTSW |
10 |
60366757 |
missense |
probably damaging |
1.00 |
|
R0082:Cdh23
|
UTSW |
10 |
60148366 |
missense |
probably damaging |
1.00 |
|
R0124:Cdh23
|
UTSW |
10 |
60143835 |
nonsense |
probably null |
|
|
R0172:Cdh23
|
UTSW |
10 |
60155411 |
missense |
probably damaging |
1.00 |
|
R0195:Cdh23
|
UTSW |
10 |
60152838 |
missense |
probably damaging |
0.99 |
|
R0365:Cdh23
|
UTSW |
10 |
60215094 |
missense |
probably damaging |
0.99 |
|
R0437:Cdh23
|
UTSW |
10 |
60246576 |
missense |
probably damaging |
1.00 |
|
R0486:Cdh23
|
UTSW |
10 |
60222725 |
missense |
probably damaging |
1.00 |
|
R0494:Cdh23
|
UTSW |
10 |
60152375 |
splice site |
probably benign |
|
|
R0545:Cdh23
|
UTSW |
10 |
60167070 |
missense |
probably benign |
0.06 |
|
R0619:Cdh23
|
UTSW |
10 |
60269556 |
missense |
probably damaging |
1.00 |
|
R0647:Cdh23
|
UTSW |
10 |
60159153 |
nonsense |
probably null |
|
|
R0647:Cdh23
|
UTSW |
10 |
60143681 |
missense |
probably damaging |
0.99 |
|
R0730:Cdh23
|
UTSW |
10 |
60159493 |
missense |
probably damaging |
0.99 |
|
R0880:Cdh23
|
UTSW |
10 |
60242200 |
missense |
possibly damaging |
0.51 |
|
R0942:Cdh23
|
UTSW |
10 |
60246639 |
missense |
possibly damaging |
0.67 |
|
R0989:Cdh23
|
UTSW |
10 |
60370289 |
missense |
probably damaging |
0.99 |
|
R1017:Cdh23
|
UTSW |
10 |
60167572 |
missense |
probably damaging |
1.00 |
|
R1173:Cdh23
|
UTSW |
10 |
60148171 |
splice site |
probably benign |
|
|
R1449:Cdh23
|
UTSW |
10 |
60212730 |
missense |
probably damaging |
1.00 |
|
R1456:Cdh23
|
UTSW |
10 |
60322899 |
missense |
possibly damaging |
0.84 |
|
R1519:Cdh23
|
UTSW |
10 |
60215122 |
missense |
possibly damaging |
0.92 |
|
R1532:Cdh23
|
UTSW |
10 |
60150110 |
missense |
probably damaging |
0.99 |
|
R1559:Cdh23
|
UTSW |
10 |
60255478 |
splice site |
probably benign |
|
|
R1704:Cdh23
|
UTSW |
10 |
60150390 |
missense |
probably damaging |
1.00 |
|
R1711:Cdh23
|
UTSW |
10 |
60359315 |
missense |
probably benign |
0.07 |
|
R1760:Cdh23
|
UTSW |
10 |
60161855 |
missense |
probably damaging |
1.00 |
|
R1782:Cdh23
|
UTSW |
10 |
60324321 |
missense |
probably damaging |
1.00 |
|
R1791:Cdh23
|
UTSW |
10 |
60227505 |
missense |
possibly damaging |
0.89 |
|
R1803:Cdh23
|
UTSW |
10 |
60167060 |
missense |
probably damaging |
1.00 |
|
R1857:Cdh23
|
UTSW |
10 |
60159076 |
missense |
probably damaging |
1.00 |
|
R1874:Cdh23
|
UTSW |
10 |
60272597 |
missense |
possibly damaging |
0.52 |
|
R1914:Cdh23
|
UTSW |
10 |
60159349 |
missense |
probably damaging |
0.99 |
|
R1958:Cdh23
|
UTSW |
10 |
60246652 |
missense |
probably benign |
0.02 |
|
R1964:Cdh23
|
UTSW |
10 |
60221001 |
missense |
probably benign |
0.31 |
|
R1966:Cdh23
|
UTSW |
10 |
60159361 |
missense |
probably damaging |
1.00 |
|
R1981:Cdh23
|
UTSW |
10 |
60214530 |
missense |
probably damaging |
1.00 |
|
R2010:Cdh23
|
UTSW |
10 |
60150006 |
missense |
probably damaging |
0.99 |
|
R2036:Cdh23
|
UTSW |
10 |
60301822 |
missense |
possibly damaging |
0.52 |
|
R2038:Cdh23
|
UTSW |
10 |
60148366 |
missense |
probably damaging |
1.00 |
|
R2044:Cdh23
|
UTSW |
10 |
60432509 |
missense |
possibly damaging |
0.72 |
|
R2111:Cdh23
|
UTSW |
10 |
60141362 |
missense |
probably damaging |
0.99 |
|
R2112:Cdh23
|
UTSW |
10 |
60141362 |
missense |
probably damaging |
0.99 |
|
R2211:Cdh23
|
UTSW |
10 |
60301783 |
missense |
possibly damaging |
0.92 |
|
R2261:Cdh23
|
UTSW |
10 |
60152907 |
missense |
probably damaging |
1.00 |
|
R2262:Cdh23
|
UTSW |
10 |
60152907 |
missense |
probably damaging |
1.00 |
|
R2306:Cdh23
|
UTSW |
10 |
60159224 |
missense |
probably damaging |
1.00 |
|
R2344:Cdh23
|
UTSW |
10 |
60152503 |
missense |
probably damaging |
1.00 |
|
R2857:Cdh23
|
UTSW |
10 |
60218432 |
critical splice donor site |
probably null |
|
|
R2858:Cdh23
|
UTSW |
10 |
60218432 |
critical splice donor site |
probably null |
|
|
R2859:Cdh23
|
UTSW |
10 |
60218432 |
critical splice donor site |
probably null |
|
|
R2876:Cdh23
|
UTSW |
10 |
60143275 |
missense |
probably damaging |
1.00 |
|
R3034:Cdh23
|
UTSW |
10 |
60244789 |
splice site |
probably benign |
|
|
R3424:Cdh23
|
UTSW |
10 |
60212660 |
missense |
possibly damaging |
0.76 |
|
R3699:Cdh23
|
UTSW |
10 |
60163149 |
critical splice donor site |
probably null |
|
|
R3700:Cdh23
|
UTSW |
10 |
60163149 |
critical splice donor site |
probably null |
|
|
R3950:Cdh23
|
UTSW |
10 |
60493105 |
missense |
probably benign |
0.04 |
|
R3951:Cdh23
|
UTSW |
10 |
60493105 |
missense |
probably benign |
0.04 |
|
R3952:Cdh23
|
UTSW |
10 |
60493105 |
missense |
probably benign |
0.04 |
|
R4108:Cdh23
|
UTSW |
10 |
60246601 |
missense |
possibly damaging |
0.51 |
|
R4114:Cdh23
|
UTSW |
10 |
60256819 |
splice site |
probably null |
|
|
R4273:Cdh23
|
UTSW |
10 |
60146940 |
missense |
possibly damaging |
0.69 |
|
R4284:Cdh23
|
UTSW |
10 |
60139272 |
missense |
possibly damaging |
0.91 |
|
R4334:Cdh23
|
UTSW |
10 |
60220838 |
missense |
probably damaging |
0.99 |
|
R4474:Cdh23
|
UTSW |
10 |
60146865 |
missense |
probably damaging |
1.00 |
|
R4532:Cdh23
|
UTSW |
10 |
60370202 |
missense |
probably benign |
0.32 |
|
R4597:Cdh23
|
UTSW |
10 |
60244823 |
missense |
probably damaging |
1.00 |
|
R4604:Cdh23
|
UTSW |
10 |
60173445 |
missense |
possibly damaging |
0.93 |
|
R4793:Cdh23
|
UTSW |
10 |
60167129 |
missense |
probably damaging |
1.00 |
|
R4816:Cdh23
|
UTSW |
10 |
60244856 |
missense |
possibly damaging |
0.93 |
|
R4833:Cdh23
|
UTSW |
10 |
60220817 |
missense |
probably damaging |
1.00 |
|
R4840:Cdh23
|
UTSW |
10 |
60255556 |
missense |
possibly damaging |
0.53 |
|
R4857:Cdh23
|
UTSW |
10 |
60227563 |
missense |
probably damaging |
1.00 |
|
R4869:Cdh23
|
UTSW |
10 |
60212713 |
missense |
probably damaging |
1.00 |
|
R4894:Cdh23
|
UTSW |
10 |
60173630 |
missense |
probably benign |
0.04 |
|
R4940:Cdh23
|
UTSW |
10 |
60143714 |
missense |
probably damaging |
0.98 |
|
R5020:Cdh23
|
UTSW |
10 |
60143811 |
missense |
probably damaging |
0.99 |
|
R5026:Cdh23
|
UTSW |
10 |
60140627 |
missense |
possibly damaging |
0.88 |
|
R5081:Cdh23
|
UTSW |
10 |
60272586 |
missense |
possibly damaging |
0.89 |
|
R5138:Cdh23
|
UTSW |
10 |
60148061 |
missense |
probably damaging |
1.00 |
|
R5236:Cdh23
|
UTSW |
10 |
60148351 |
missense |
probably damaging |
1.00 |
|
R5361:Cdh23
|
UTSW |
10 |
60493044 |
critical splice donor site |
probably null |
|
|
R5384:Cdh23
|
UTSW |
10 |
60173541 |
missense |
probably damaging |
0.99 |
|
R5500:Cdh23
|
UTSW |
10 |
60150090 |
missense |
probably damaging |
1.00 |
|
R5512:Cdh23
|
UTSW |
10 |
60370165 |
splice site |
probably null |
|
|
R5673:Cdh23
|
UTSW |
10 |
60143636 |
missense |
probably damaging |
1.00 |
|
R5720:Cdh23
|
UTSW |
10 |
60228802 |
missense |
possibly damaging |
0.71 |
|
R5726:Cdh23
|
UTSW |
10 |
60243259 |
missense |
probably damaging |
0.98 |
|
R5732:Cdh23
|
UTSW |
10 |
60167096 |
missense |
possibly damaging |
0.80 |
|
R5739:Cdh23
|
UTSW |
10 |
60141388 |
missense |
probably damaging |
0.99 |
|
R5760:Cdh23
|
UTSW |
10 |
60242171 |
missense |
probably damaging |
0.99 |
|
R5793:Cdh23
|
UTSW |
10 |
60141907 |
missense |
probably damaging |
1.00 |
|
R5880:Cdh23
|
UTSW |
10 |
60220713 |
missense |
probably damaging |
1.00 |
|
R5905:Cdh23
|
UTSW |
10 |
60370314 |
missense |
probably damaging |
0.98 |
|
R5907:Cdh23
|
UTSW |
10 |
60264158 |
missense |
probably damaging |
1.00 |
|
R5910:Cdh23
|
UTSW |
10 |
60213600 |
missense |
possibly damaging |
0.81 |
|
R5932:Cdh23
|
UTSW |
10 |
60228763 |
missense |
probably damaging |
1.00 |
|
R5996:Cdh23
|
UTSW |
10 |
60249356 |
missense |
possibly damaging |
0.85 |
|
R6015:Cdh23
|
UTSW |
10 |
60143761 |
missense |
probably damaging |
0.97 |
|
R6020:Cdh23
|
UTSW |
10 |
60167105 |
missense |
probably damaging |
1.00 |
|
R6023:Cdh23
|
UTSW |
10 |
60301321 |
missense |
probably damaging |
1.00 |
|
R6028:Cdh23
|
UTSW |
10 |
60370314 |
missense |
probably damaging |
0.98 |
|
R6066:Cdh23
|
UTSW |
10 |
60269537 |
missense |
probably damaging |
1.00 |
|
R6137:Cdh23
|
UTSW |
10 |
60270291 |
missense |
probably damaging |
0.96 |
|
R6211:Cdh23
|
UTSW |
10 |
60246600 |
missense |
possibly damaging |
0.90 |
|
R6298:Cdh23
|
UTSW |
10 |
60262451 |
nonsense |
probably null |
|
|
R6302:Cdh23
|
UTSW |
10 |
60140872 |
missense |
possibly damaging |
0.74 |
|
R6338:Cdh23
|
UTSW |
10 |
60248930 |
missense |
probably damaging |
1.00 |
|
R6356:Cdh23
|
UTSW |
10 |
60274626 |
missense |
probably damaging |
1.00 |
|
R6441:Cdh23
|
UTSW |
10 |
60143815 |
missense |
probably damaging |
1.00 |
|
R6714:Cdh23
|
UTSW |
10 |
60167609 |
missense |
possibly damaging |
0.62 |
|
R6760:Cdh23
|
UTSW |
10 |
60141947 |
missense |
probably damaging |
1.00 |
|
R6807:Cdh23
|
UTSW |
10 |
60214650 |
missense |
possibly damaging |
0.95 |
|
R6855:Cdh23
|
UTSW |
10 |
60141901 |
missense |
possibly damaging |
0.66 |
|
R6937:Cdh23
|
UTSW |
10 |
60322893 |
missense |
probably damaging |
1.00 |
|
R6942:Cdh23
|
UTSW |
10 |
60274635 |
missense |
possibly damaging |
0.93 |
|
R6961:Cdh23
|
UTSW |
10 |
60485893 |
missense |
probably benign |
0.00 |
|
R7009:Cdh23
|
UTSW |
10 |
60173085 |
missense |
probably damaging |
0.99 |
|
R7010:Cdh23
|
UTSW |
10 |
60366770 |
missense |
probably benign |
0.03 |
|
R7032:Cdh23
|
UTSW |
10 |
60167567 |
missense |
probably damaging |
1.00 |
|
R7046:Cdh23
|
UTSW |
10 |
60214530 |
missense |
probably damaging |
1.00 |
|
R7111:Cdh23
|
UTSW |
10 |
60222823 |
missense |
probably damaging |
1.00 |
|
R7196:Cdh23
|
UTSW |
10 |
60143759 |
missense |
probably damaging |
0.99 |
|
R7198:Cdh23
|
UTSW |
10 |
60148378 |
missense |
possibly damaging |
0.91 |
|
R7223:Cdh23
|
UTSW |
10 |
60167596 |
missense |
probably damaging |
1.00 |
|
R7290:Cdh23
|
UTSW |
10 |
60212620 |
missense |
probably benign |
|
|
R7335:Cdh23
|
UTSW |
10 |
60140895 |
missense |
probably damaging |
1.00 |
|
R7340:Cdh23
|
UTSW |
10 |
60366775 |
missense |
probably benign |
0.19 |
|
R7350:Cdh23
|
UTSW |
10 |
60246689 |
missense |
probably damaging |
1.00 |
|
R7366:Cdh23
|
UTSW |
10 |
60151471 |
nonsense |
probably null |
|
|
R7374:Cdh23
|
UTSW |
10 |
60153679 |
missense |
probably damaging |
0.99 |
|
R7455:Cdh23
|
UTSW |
10 |
60142003 |
missense |
possibly damaging |
0.82 |
|
R7537:Cdh23
|
UTSW |
10 |
60220724 |
missense |
probably benign |
0.17 |
|
R7573:Cdh23
|
UTSW |
10 |
60159329 |
missense |
probably benign |
0.17 |
|
R7578:Cdh23
|
UTSW |
10 |
60243186 |
missense |
probably benign |
0.14 |
|
R7646:Cdh23
|
UTSW |
10 |
60140931 |
missense |
possibly damaging |
0.95 |
|
R7703:Cdh23
|
UTSW |
10 |
60173043 |
missense |
probably damaging |
1.00 |
|
R7763:Cdh23
|
UTSW |
10 |
60148356 |
missense |
probably damaging |
1.00 |
|
R7797:Cdh23
|
UTSW |
10 |
60220973 |
missense |
probably benign |
0.07 |
|
R7867:Cdh23
|
UTSW |
10 |
60150390 |
missense |
probably damaging |
1.00 |
|
R7878:Cdh23
|
UTSW |
10 |
60149979 |
missense |
possibly damaging |
0.69 |
|
R7915:Cdh23
|
UTSW |
10 |
60143668 |
missense |
probably damaging |
0.97 |
|
R7922:Cdh23
|
UTSW |
10 |
60218485 |
missense |
probably benign |
0.31 |
|
R7963:Cdh23
|
UTSW |
10 |
60171967 |
missense |
probably damaging |
1.00 |
|
R7997:Cdh23
|
UTSW |
10 |
60432518 |
missense |
possibly damaging |
0.81 |
|
R8167:Cdh23
|
UTSW |
10 |
60150162 |
missense |
probably benign |
0.12 |
|
R8167:Cdh23
|
UTSW |
10 |
60173472 |
missense |
probably damaging |
0.96 |
|
R8258:Cdh23
|
UTSW |
10 |
60151435 |
missense |
probably damaging |
0.99 |
|
R8259:Cdh23
|
UTSW |
10 |
60151435 |
missense |
probably damaging |
0.99 |
|
R8317:Cdh23
|
UTSW |
10 |
60272568 |
missense |
probably damaging |
1.00 |
|
R8317:Cdh23
|
UTSW |
10 |
60147037 |
critical splice donor site |
probably null |
|
|
R8326:Cdh23
|
UTSW |
10 |
60274591 |
missense |
possibly damaging |
0.55 |
|
R8333:Cdh23
|
UTSW |
10 |
60150390 |
missense |
probably damaging |
1.00 |
|
R8348:Cdh23
|
UTSW |
10 |
60167507 |
missense |
probably benign |
0.43 |
|
R8366:Cdh23
|
UTSW |
10 |
60160799 |
missense |
probably benign |
|
|
R8504:Cdh23
|
UTSW |
10 |
60274618 |
missense |
probably benign |
0.00 |
|
R8676:Cdh23
|
UTSW |
10 |
60246689 |
missense |
probably damaging |
1.00 |
|
R8781:Cdh23
|
UTSW |
10 |
60167567 |
missense |
probably damaging |
1.00 |
|
R8785:Cdh23
|
UTSW |
10 |
60147114 |
missense |
probably damaging |
1.00 |
|
R8788:Cdh23
|
UTSW |
10 |
60324372 |
missense |
probably damaging |
1.00 |
|
R8802:Cdh23
|
UTSW |
10 |
60244877 |
missense |
probably benign |
0.04 |
|
R8837:Cdh23
|
UTSW |
10 |
60160755 |
missense |
probably benign |
0.28 |
|
R8863:Cdh23
|
UTSW |
10 |
60212613 |
nonsense |
probably null |
|
|
R8889:Cdh23
|
UTSW |
10 |
60143284 |
missense |
probably damaging |
0.97 |
|
R8892:Cdh23
|
UTSW |
10 |
60143284 |
missense |
probably damaging |
0.97 |
|
R8921:Cdh23
|
UTSW |
10 |
60140908 |
missense |
probably damaging |
0.99 |
|
R8980:Cdh23
|
UTSW |
10 |
60173625 |
missense |
probably benign |
0.06 |
|
R9000:Cdh23
|
UTSW |
10 |
60140277 |
missense |
possibly damaging |
0.82 |
|
R9043:Cdh23
|
UTSW |
10 |
60151478 |
missense |
probably benign |
0.00 |
|
R9046:Cdh23
|
UTSW |
10 |
60218303 |
intron |
probably benign |
|
|
R9070:Cdh23
|
UTSW |
10 |
60173539 |
missense |
probably benign |
|
|
R9075:Cdh23
|
UTSW |
10 |
60153541 |
missense |
probably damaging |
1.00 |
|
R9132:Cdh23
|
UTSW |
10 |
60270283 |
splice site |
probably benign |
|
|
R9155:Cdh23
|
UTSW |
10 |
60249485 |
missense |
probably damaging |
0.99 |
|
R9171:Cdh23
|
UTSW |
10 |
60161810 |
missense |
probably benign |
0.00 |
|
R9179:Cdh23
|
UTSW |
10 |
60153664 |
missense |
probably benign |
0.06 |
|
R9186:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9189:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9207:Cdh23
|
UTSW |
10 |
60243210 |
missense |
probably damaging |
1.00 |
|
R9240:Cdh23
|
UTSW |
10 |
60215044 |
missense |
probably benign |
0.00 |
|
R9244:Cdh23
|
UTSW |
10 |
60249442 |
missense |
possibly damaging |
0.93 |
|
R9284:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9286:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9287:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9302:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9352:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9353:Cdh23
|
UTSW |
10 |
60143306 |
missense |
possibly damaging |
0.54 |
|
R9423:Cdh23
|
UTSW |
10 |
60148387 |
missense |
probably damaging |
1.00 |
|
R9513:Cdh23
|
UTSW |
10 |
60166995 |
missense |
probably damaging |
0.99 |
|
R9577:Cdh23
|
UTSW |
10 |
60146895 |
missense |
probably damaging |
1.00 |
|
R9598:Cdh23
|
UTSW |
10 |
60214574 |
missense |
probably benign |
0.01 |
|
R9631:Cdh23
|
UTSW |
10 |
60243168 |
missense |
possibly damaging |
0.49 |
|
R9652:Cdh23
|
UTSW |
10 |
60167135 |
missense |
probably damaging |
1.00 |
|
R9725:Cdh23
|
UTSW |
10 |
60432561 |
missense |
probably benign |
0.02 |
|
X0052:Cdh23
|
UTSW |
10 |
60220913 |
missense |
probably damaging |
1.00 |
|
Z1088:Cdh23
|
UTSW |
10 |
60249423 |
missense |
probably benign |
0.35 |
|
Z1176:Cdh23
|
UTSW |
10 |
60264100 |
missense |
probably benign |
|
|
Z1176:Cdh23
|
UTSW |
10 |
60146549 |
missense |
probably damaging |
1.00 |
|
Z1177:Cdh23
|
UTSW |
10 |
60270393 |
critical splice acceptor site |
probably null |
|
|
Z1177:Cdh23
|
UTSW |
10 |
60159334 |
missense |
possibly damaging |
0.80 |
|
|---|
| Mode of Inheritance |
Autosomal Recessive |
| Local Stock | Live Mice |
|---|
| MMRRC Submission |
036997-MU
|
|---|
| Last Updated |
2018-10-11 12:30 PM
by Diantha La Vine
|
| Record Created |
2013-04-11 6:07 PM
by Adam Dismang
|
| Record Posted |
2014-03-28 |
|---|
| Phenotypic Description |
The dee dee mutation was induced by N-ethyl-N-nitrosourea (ENU) mutagenesis and was discovered in G3 animals. Homozygous mutant mice exhibit excessive head bobbing.
|
|---|
| Nature of Mutation |  The mice in the dee_dee pedigree were genotyped and the causative mutation for the head bobbing phenotype was mapped using quickmapping. Among six mice with the dee dee phenotype, five were homozygous and one was heterozygous for a mutation in Cdh23 on chromosome 10. Among 19 unaffected mice, all were either heterozygous or wild type at the Cdh23 locus, resulting in a LOD score of 5.618 for the Cdh23 mutation. The mutation is a C to A transversion at base pair 60308056 (v38) on Chromosome 10, equivalent to base pair 388435 in the GenBank genomic region NC_000076 encoding Cdh23. The mutation corresponds to residue 9176 in the NM_023370.3 mRNA sequence in exon 62 of 71 total exons. The mutation also corresponds to residue 9710 in the NM_001252635 mRNA sequence in exon 61 of 70 total exons. The mice in the dee_dee pedigree were genotyped and the causative mutation for the head bobbing phenotype was mapped using quickmapping. Among six mice with the dee dee phenotype, five were homozygous and one was heterozygous for a mutation in Cdh23 on chromosome 10. Among 19 unaffected mice, all were either heterozygous or wild type at the Cdh23 locus, resulting in a LOD score of 5.618 for the Cdh23 mutation. The mutation is a C to A transversion at base pair 60308056 (v38) on Chromosome 10, equivalent to base pair 388435 in the GenBank genomic region NC_000076 encoding Cdh23. The mutation corresponds to residue 9176 in the NM_023370.3 mRNA sequence in exon 62 of 71 total exons. The mutation also corresponds to residue 9710 in the NM_001252635 mRNA sequence in exon 61 of 70 total exons.
The genomic DNA sequence is shown; numbering corresponds to NC_000076. The mutated nucleotide is indicated in red. The mutation results in a tyrosine (Y) to premature stop codon (*) substitution at amino acid residue 2923 in ENSMUSP00000101101 or residue 2921 in ENSMUSP00000101104.
|
|---|
Illustration of Mutations in
Gene & Protein |
|
|---|
| Protein Prediction |
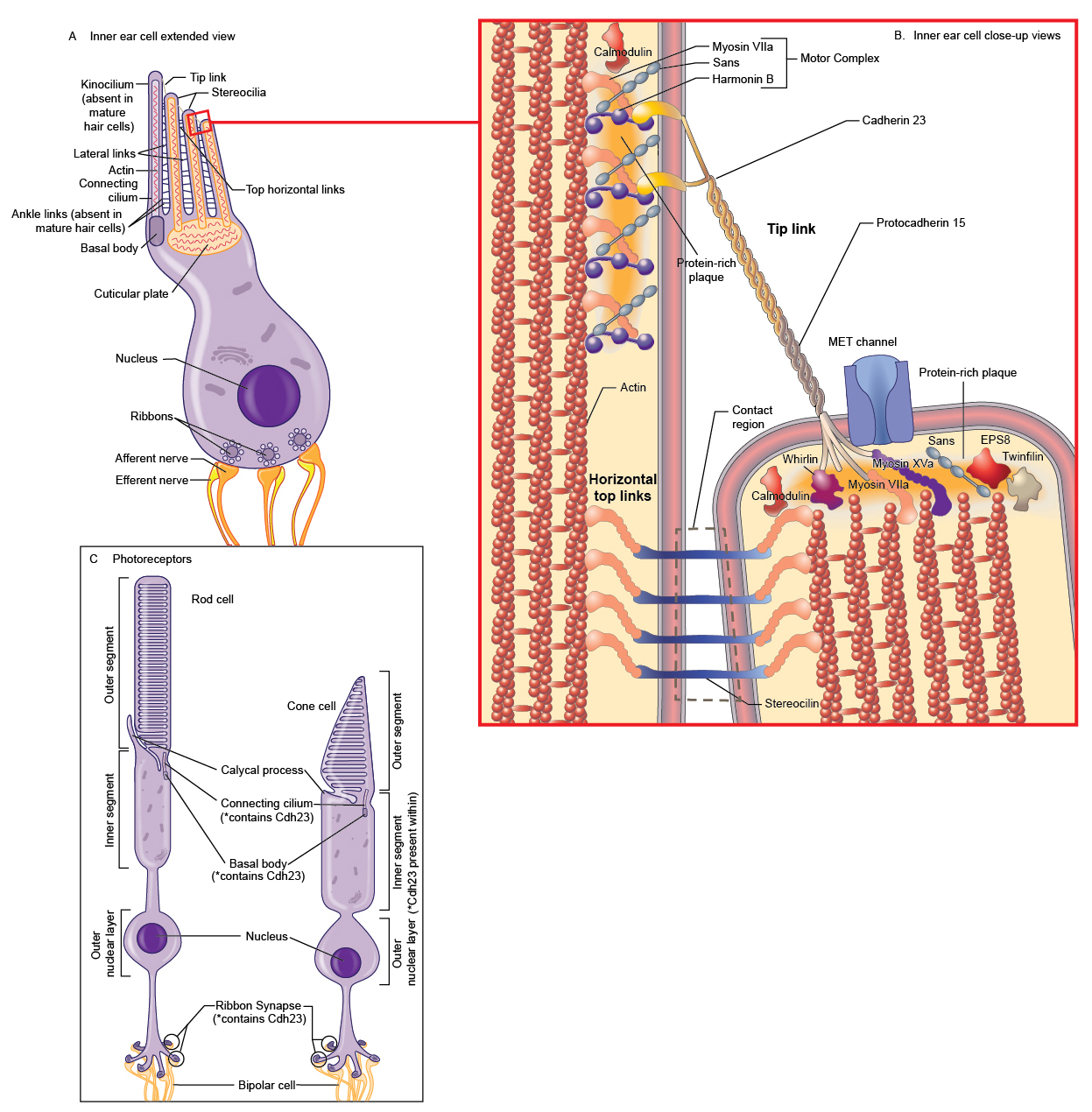
Cdh23 (alternatively, Ush1d) encodes cadherin 23 (Cdh23), a member of the cadherin superfamily of cell-cell adhesion proteins (1). The cadherin proteins share similar features including a signal peptide (amino acids (aa) 1-23 in Cdh23), an extracellular domain (aa 24-3064 in Cdh23) with a variable number of extracellular ectodomain (EC) repeats (alternatively, cadherin motifs; Cdh23 has 27 EC repeats), a single helical transmembrane domain (aa 3065-3085 in Cdh23), and a cytoplasmic C-terminus (aa 3086-3354 in Cdh23) that connects the cadherin protein to the actin cytoskeleton through interactions with α- and β-catenin (Figure 1; (1-4)). The EC repeats of Cdh23 are each approximately 108 aa long and are 41%-57% similar to the ectodomains of other cadherins (e.g., Cdh1, 33-44%; Cdh2, 37-41%) (5). Similar to other members of the cadherin family (6), each Cdh23 EC repeat has four calcium binding motifs that together bind three calcium ions. Calcium binding facilitates linearization and rigidification of the cadherin ectodomain, prevents ectodomain unfolding, and promotes cadherin dimerization and consequent cell-cell adhesion (5;7;8). The overall 3D structure of the cadherin repeat motif is quite similar to the Greek-key topology of immunoglobulin domains with seven β-strands arranged as two opposing β-sheets with N- and C-termini at the opposite ends (2;7). Cdh23 forms homodimers and heterodimers with protocadherin 15 (Pcdh15; see the record for squirm) through interactions along the full length of the extracellular domain (9;10). Cdh23 interacts with Pcdh15 to form tip links, extracellular filaments that connect the tips of hair cell stereocilia and function in the gating of the hair cell’s mechanotransducer channel (see “Background” for more details; (8;9;11;12)). A crystal structure of EC1 and EC2 from mouse Cdh23 and EC1 and EC2 from mouse Pcdh15 determined that Cdh23 and Pcdh15 form an overlapped, antiparallel heterodimer through two main sites of interaction [Figure 2; PDB: 4AQ8; (10)]. The first is Leu145 and Gln187 in Cdh23 with Tyr8, Pro19, and Ile108 in Pcdh15 and the second involves Tyr16 and Gln98 of Cdh23 with Ile122, Arg113, and Val115 in Pcdh15 (10). Cdh23 forms a complex with several proteins to gate the mechanoelectrical transduction channel, to form the stereocilia bundle, and to maintain the tip link in hair cells [Table 1; (13-15)]. Within the cytoplasmic domain of Cdh23 are two PDZ-binding interfaces (PBIs) that interact with PDZ domains of other proteins. For example, the internal PBI (aa 3177-3270) of Cdh23 interacts with PDZ1 of harmonin, while the C-terminal PBI of Cdh23 (aa 3351-3354) interacts with PDZ2 of harmonin as well as with MAGI-1 (16;17). Table 1. Cdh23-interacting proteins
|
Interacting protein (synonym)
|
Brief Description
|
Cdh23-associated function
|
Refs
|
|
Pcdh15 (USH1F)
|
See the record for squirm
|
Pcdh15 homodimers interact with Cdh23 homodimers (alternatively, Cdh23 and Pcdh15 form a heterodimer) to form tip links, which gate the mechanoelectrical transduction channel of hair cells
|
(9)
|
|
Harmonin (USH1C)
|
Scaffold protein
|
Harmonin binds to Cdh23, Pcdh15, and F-actin to achor the interstereocilia links to the stereocilia actin core
|
(13;16;18;19)
|
|
Myo7a (USH1B)
|
Actin-based molecular motor
|
Interacts with Cdh23 and harmonin to facilitate the cohesion of the stereocilia
|
(18;20;21)
|
|
Myo1c
|
Actin-based molecular motor
|
Forms a complex with Cdh23 in the mechanotransduction apparatus
|
(22)
|
|
MAGI-1
|
Scaffold protein
|
Interacts with Cdh23 beneath the stereociliary plasma membrane to connect the cytoskeleton with the Cdh23-harmonin-Myo7a complex
|
(17)
|
|
SANS (USH1G)
|
Scaffold protein
|
Interacts with Cdh23, Myo7a and harmonin complex to maintain the tip link
|
(23;24)
|
|
EHD4
|
Endocytic recycling
|
May regulate calcium-sensitive Cdh23 trafficking/localization in cochlear hair cells
|
(25)
|
Please see the record for squirm for more information about the cadherin proteins. Alternative splicing of Cdh23 in both mice and humans involves the inclusion [Cdh23(+68)] or exclusion [Cdh23(-68)] of exon 68, which encodes 35 cytoplasmic amino acids that when present, interrupt the internal PBI of Cdh23 (3-5;16;26). Harmonin binding to the hair cell-specific Cdh23(+68) isoform is reduced compared to binding to the Cdh23(-68) isoform, which is prevalent in the retina (16). Lagziel et al. described the identification of two Cdh23 isoforms (B1 and B2) of differing lengths (3787 bp (AY563163) and 3682 bp (AY563164) in mouse, respectively; 4044 bp and 3939 bp in human, respectively) (Figure 1; (26)). The B isoforms are predicted to encode proteins with seven EC motifs, a single transmembrane domain, and a cytoplasmic domain with (B1) or without (B2) the amino acid sequence encoded by exon 68 (26). Two additional isoforms were identified and are 1783 bp (C1; exons 66-69) and 1678 bp (C2; exons 66, 67, and 69) in humans (26). In mice, the C1 and C2 isoforms are encoded by the corresponding exons and are 1172 bp (AY563159) and 1064 bp (AY563160), respectively. The C1 and C2 isoforms encode the cytoplasmic domain of Cdh23 and do not have transmembrane domains, EC repeats, or a signal peptide; C1 and C2 are expected to be cytosolic (13;26). The mouse and human C isoforms have a novel exonic sequence, 65a, encoding the seven N-terminal amino acids of these proteins (MLLPNYR) (26). Exon 65a is located immediately upstream of the 5’ boundary of exon 66 in full-length Cdh23 (13;26). Michel et al. propose that the C1 and C2 isoforms mediate the interaction of the transmembrane forms of Cdh23 with cytosolic binding partners (13). The dee dee mutation encodes a premature stop codon within EC27. The mutation is predicted to affect all of the Cdh23 isoforms with the exception of C1 and C2, although expression of the Cdh23 isoforms has not been examined.
|
|---|
| Expression/Localization | Reverse-transcriptase (RT)-PCR detected Cdh23 in several adult tissues in the mouse including brain, heart, kidney, liver, spleen, ear, skeletal muscle, eye, thymus, lung, and testis (1;27). In the mouse embryo, Cdh23 is expressed as early as embryonic day (E) 7 (27). At E18, in situ hybridization detected high Cdh23 expression in the components of the vestibular system (i.e., semicircular canals, saccule, utricle, and cochlea of the inner ear) that maintains balance and equilibrium (27). Within the mouse inner ear, Cdh23 is highly expressed in sensory hair cells and Reissner’s membrane (27). At postnatal day (P)7, P9, and P12, Cdh23 protein expression was detected in the cochlea and vestibule in the inner ear as well as in the eye, brain and testis of the mouse (28). In hair cells of the inner ear, Cdh23 is localized to the centrosome, transient lateral links, tip links, and kinociliary links that link the kinocilium (the genuine cilium in the immature hair bundle) to the adjacent stereocilia; Cdh23 is also detected in Reissner’s membrane (1;3;9;13;16;18;25;26). Upon hair cell maturation, Cdh23 becomes restricted to the distal tip of the hair bundle (13). Northern blot of human RNA detected CDH23 in the retina; expression was not detected in the ciliary body, retinal pigmented epithelium (RPE)-choroid, lens, iris, brain, heart, placenta, lung, liver, skeletal muscle, or kidney (3). No explanation was offered for the differences observed between the expression pattern of mouse and human Cdh23. Northern blot of macula and peripheral retina total RNA from monkey detected Cdh23 RNA in both retinal regions (3). Within the retina, Cdh23 is localized to both rod and cone photoreceptors in the inner segment, the connecting cilium, the basal body complex, and the ribbon synapses; Cdh23 is not expressed in the RPE [(29); reviewed in (30)]. Apostolopoulou et al. propose that Cdh23 is upregulated in human breast cancer (31). Cdh23 is localized to homotypic adhesions between MCF-7 cells (a breast cancer epithelial cell line) as well as to heterotypic contacts between MCF-7 cells and normal breast fibroblasts (NBFs) after co-culture of MCF-7 cells and NBFs (31). In NBF cells, Cdh23 is also localized to centrosomes (31). In tumor and healthy breast tissue samples, Cdh23 is more highly expressed in luminal epithelia than in stroma (31). Cdh23(+68) is the exclusive isoform expressed by hair cells and is predominantly (if not exclusively) expressed in organ of Corti within the inner ear of the mouse (5;16;22). Cdh23(-68) is expressed in the cochlea and vestibule of the inner ear as well as in the eye, brain and testis of the mouse (28). Both the transmembrane (B1 and B2) and cytosolic (C1 and C2) forms of Cdh23 are expressed in the cochlea; additional expression analysis on these isoforms has not been documented (13). The cytosolic isoforms are localized to the centrosome (13;26).
|
|---|
| Background |
Cell-cell adhesive connections mediated by cadherins function in diverse processes including cell sorting, synapse formation, the development and function of sensory cells in the inner ear and retina, and cell migration during development and in the adult [Figure 3; (32); reviewed in (33)]. At their apical surface, hair cells in the cochlea of the inner ear have a single kinocilium and a bundle of actin-filled stereocilia arranged in a staircase of increasing height (18;34;35). Each stereocilium is connected to its taller neighbor by the tip link (36); side links, ankle links, and horizontal top connectors also connect the individual stereocilia and coordinate their movement (33;37). During deflection of the hair bundle, the tip link translates the mechanical forces arising from sound waves and head movement into electrochemical signals through gating of the mechanoelectric transducer channels at the lower end of each tip link (17;34;37); these electrical signals facilitate hearing and balance (8). Cdh23 interacts with Pcdh15 to form the tip link (5;28;38). In the synaptic terminals of the photoreceptor cells of the retina, Cdh23 colocalizes with Pcdh15, harmonin, and Myo7a. The complex in the photoreceptor synapse is proposed to have a role in the structural and functional organization of the synaptic junction (39). See the record squirm for more information about the sensory cells in the inner ear and retina as well as the known functions of Cdh23, Pcdh15, and the Usher syndrome-associated proteins therein. In humans, missense mutations in CDH23 are typically linked to autosomal recessive deafness 12 [DFNB12; OMIM: #601386; (3;40)], while null mutations are typically linked to Usher syndrome type 1D (USH1D) and Usher syndrome type 1D/F digenic [OMIM: #601067; (3;4)]. DFNB12 is a form of nonsyndromic autosomal recessive deafness. The phenotype in patients with DFNB12 can also include moderate to profound high-frequency progressive sensorineural hearing loss; vestibular and retinal function are normal in patients with DFNB12 (40). USH1D is characterized by hearing loss, retinitis pigmentosa leading to blindness, and in some cases, vestibular dysfunction (15;18;38). Type 1D/F Usher syndrome is caused by heterozygous mutations in both CDH23 and PCDH15. Patients with Usher syndrome type 1D/F exhibit congenital nonprogressive deafness, vestibular dysfunction, and retinitis pigmentosa (41). Several Cdh23 mutant mouse alleles have been reported and all (with the exception of Cdh23v-bus (MGI:1856681), a donor splice site mutation after exon 67 in Cdh23) are within or affect an EC domain (1;5;21;27;38;42-45). Similar to the mutations found in humans, nonsense and splice-site mutations that lead to Cdh23 null alleles result in USHD1 phenotypes, while missense mutations result in phenotypes characteristic of DFNB12 [reviewed in (38)]. The null mutations [e.g., waltzer (Cdh23v; MGI:1856228)] affect hair bundle development and cause defects in the organization of the stereocilial bundle in the cochlear hair cells and misplaced kinocilia (1;44;46). These defects lead to both hearing loss and vestibular defects (as exhibited by circling, head tilt, and/or head tossing) (1;43;44). In contrast, the hair cells and hair bundles develop normally in mouse mutants with missense mutations of Cdh23, but the function is affected (21). Although all of the mouse models exhibit hearing loss; retinal degeneration does not occur (38). Examination of three Cdh23 null mutants using electroretinography determined that two out of the three mutants exhibited abnormal retinal function; cone photoreceptor function was mildly attenuated as well as the response of retinal interneurons (15). However, histological examination determined that the anatomy of the retinas were normal (15). Libby et al. propose that the difference between the mouse and human retina phenotypes in null mutants is dependent on genetic background (15). It is possible that in other genetic backgrounds that have yet to be tested, Cdh23 null alleles may exhibit retinal degeneration. Cdh23 mutations in mice can also cause noise-induced and age-related hearing loss. Mice heterozygous for a Cdh23 null mutation (Cdh23v) are predisposed to noise-induced hearing loss (47). Sequence analysis of the CBA/CaJ strain (a strain with normal hearing throughout life) and the C57BL/6J strain (a strain that develops age-related hearing loss), as well as 54 additional inbred strains, determined that a single-nucleotide polymorphism, Cdh23G753A, showed significant association with age-related hearing loss (42). An ENU-induced mouse Cdh23 allele, salsa (MGI:3708379), causes progressive hearing loss due to a missense mutation predicted to affect calcium binding by the extracellular domain (21). These mice do not have hair bundle development defects or vestibular dysfunction, however, tip links are progressively lost, subsequently leading to hair cell death (21). The salsa mutation alters the interaction of Cdh23 with Pcdh15 (21). Another ENU-induced allele, erlong (MGI:4453135), contains a recessive missense mutation that leads to progressive hearing loss with no vestibular abnormalities (45). The jera model (MGI: 5140884) harbors an ENU-induced missense Cdh23 mutation that causes more severe hearing loss than that observed in salsa and erlong mice; the development of hair bundles appears normal in jera (38). The jera mouse is also susceptible to noise-induced hearing loss (38).
|
|---|
| Putative Mechanism | The dee dee mice exhibit vestibular dysfunction similar to waltzer and other Cdh23 null mouse models. Another spontaneous premature stop codon twelve amino acids away from the dee dee mutation occurs at residue 2935 (Arg2935*) in the mouse model Cdh23V-5J (MGI:1857520). Homozygous Cdh23V-5J mice exhibit head bobbing and circling and are deaf. Dee dee mice have not been examined for deafness, but share the other Cdh23V-5J and waltzer phenotypes, indicating that dee dee is probably a null mutation that results in alterations in the organization of the stereocilia bundle in hair cells of the cochlea and the vestibule as well as misplaced kinocilia.
|
|---|
| Primers |
PCR Primer
dee_dee_pcr_F: GCAGTGTTTACATAAAGCTGGAGCAAC
dee_dee_pcr_R: AGGCGAATGAACTCCTCCTCGAAG
|
|---|
| Genotyping | Dee dee genotyping is performed by amplifying the region containing the mutation using PCR, followed by sequencing of the amplified region to detect the single nucleotide transition. PCR Primers
DeeDee(F): 5’-GCAGTGTTTACATAAAGCTGGAGCAAC- 3’
DeeDee(R): 5’- AGGCGAATGAACTCCTCCTCGAAG- 3’ Sequencing Primer
DeeDee_seq(R): 5’- TCCTCCTCGAAGCCACG- 3’ PCR program
1) 94°C 2:00
2) 94°C 0:30
3) 55°C 0:30
4) 72°C 1:00
5) repeat steps (2-4) 40X
6) 72°C 10:00
7) 4°C ∞ The following sequence of 409 nucleotides (from Genbank genomic region NC_000073 for linear DNA sequence of Cdh23) is amplified: 388191 gcagtgttta
388201 cataaagctg gagcaaccac taaaaatcag gagcgtttca gccaggcatg ctagcgtatg
388261 cctataatcc cagcactcag gctgtggacg tggagaactc tgacttcctc caaggatgtg
388321 gcttaggggg cagggcttat catcagtatg tgtggcctct gagctgtgcc accccgactc
388381 tttaacaggg agtgtggacg gtatcctgcg cacctttgac ctcttcatgg cctacagccc
388441 tggctatttt gtggtagaca tcgtggcccg agacctggcc ggccacaatg ataccgccat
388501 catcggcatc tacatcctga gggatgacca gcgcgtgaag atcgtcatca atgagatccc
388561 ggaccgcgtg cgtggcttcg aggaggagtt cattcgcct Sense strand shown. Primer binding sites are underlined and the sequencing primer is highlighted; the mutated nucleotide is shown in red text (C>A, sense strand; G>T, Chr. + strand).
|
|---|
| References | 1. Di Palma, F., Holme, R. H., Bryda, E. C., Belyantseva, I. A., Pellegrino, R., Kachar, B., Steel, K. P., and Noben-Trauth, K. (2001) Mutations in Cdh23, Encoding a New Type of Cadherin, Cause Stereocilia Disorganization in Waltzer, the Mouse Model for Usher Syndrome Type 1D. Nat Genet. 27, 103-107. 3. Bork, J. M., Peters, L. M., Riazuddin, S., Bernstein, S. L., Ahmed, Z. M., Ness, S. L., Polomeno, R., Ramesh, A., Schloss, M., Srisailpathy, C. R., Wayne, S., Bellman, S., Desmukh, D., Ahmed, Z., Khan, S. N., Kaloustian, V. M., Li, X. C., Lalwani, A., Riazuddin, S., Bitner-Glindzicz, M., Nance, W. E., Liu, X. Z., Wistow, G., Smith, R. J., Griffith, A. J., Wilcox, E. R., Friedman, T. B., and Morell, R. J. (2001) Usher Syndrome 1D and Nonsyndromic Autosomal Recessive Deafness DFNB12 are Caused by Allelic Mutations of the Novel Cadherin-Like Gene CDH23. Am J Hum Genet. 68, 26-37. 4. Bolz, H., von Brederlow, B., Ramirez, A., Bryda, E. C., Kutsche, K., Nothwang, H. G., Seeliger, M., del C-Salcedo Cabrera, M., Vila, M. C., Molina, O. P., Gal, A., and Kubisch, C. (2001) Mutation of CDH23, Encoding a New Member of the Cadherin Gene Family, Causes Usher Syndrome Type 1D. Nat Genet. 27, 108-112. 7. Elledge, H. M., Kazmierczak, P., Clark, P., Joseph, J. S., Kolatkar, A., Kuhn, P., and Muller, U. (2010) Structure of the N Terminus of Cadherin 23 Reveals a New Adhesion Mechanism for a Subset of Cadherin Superfamily Members. Proc Natl Acad Sci U S A. 107, 10708-10712. 9. Kazmierczak, P., Sakaguchi, H., Tokita, J., Wilson-Kubalek, E. M., Milligan, R. A., Muller, U., and Kachar, B. (2007) Cadherin 23 and Protocadherin 15 Interact to Form Tip-Link Filaments in Sensory Hair Cells. Nature. 449, 87-91. 11. Lelli, A., Kazmierczak, P., Kawashima, Y., Muller, U., and Holt, J. R. (2010) Development and Regeneration of Sensory Transduction in Auditory Hair Cells Requires Functional Interaction between Cadherin-23 and Protocadherin-15. J Neurosci. 30, 11259-11269. 12. Alagramam, K. N., Goodyear, R. J., Geng, R., Furness, D. N., van Aken, A. F., Marcotti, W., Kros, C. J., and Richardson, G. P. (2011) Mutations in Protocadherin 15 and Cadherin 23 Affect Tip Links and Mechanotransduction in Mammalian Sensory Hair Cells. PLoS One. 6, e19183. 13. Michel, V., Goodyear, R. J., Weil, D., Marcotti, W., Perfettini, I., Wolfrum, U., Kros, C. J., Richardson, G. P., and Petit, C. (2005) Cadherin 23 is a Component of the Transient Lateral Links in the Developing Hair Bundles of Cochlear Sensory Cells. Dev Biol. 280, 281-294. 14. Yonezawa, S., Hanai, A., Mutoh, N., Moriyama, A., and Kageyama, T. (2008) Redox-Dependent Structural Ambivalence of the Cytoplasmic Domain in the Inner Ear-Specific Cadherin 23 Isoform. Biochem Biophys Res Commun. 366, 92-97. 15. Libby, R. T., Kitamoto, J., Holme, R. H., Williams, D. S., and Steel, K. P. (2003) Cdh23 Mutations in the Mouse are Associated with Retinal Dysfunction but Not Retinal Degeneration. Exp Eye Res. 77, 731-739. 16. Siemens, J., Kazmierczak, P., Reynolds, A., Sticker, M., Littlewood-Evans, A., and Muller, U. (2002) The Usher Syndrome Proteins Cadherin 23 and Harmonin Form a Complex by Means of PDZ-Domain Interactions. Proc Natl Acad Sci U S A. 99, 14946-14951. 17. Xu, Z., Peng, A. W., Oshima, K., and Heller, S. (2008) MAGI-1, a Candidate Stereociliary Scaffolding Protein, Associates with the Tip-Link Component Cadherin 23. J Neurosci. 28, 11269-11276. 18. Boeda, B., El-Amraoui, A., Bahloul, A., Goodyear, R., Daviet, L., Blanchard, S., Perfettini, I., Fath, K. R., Shorte, S., Reiners, J., Houdusse, A., Legrain, P., Wolfrum, U., Richardson, G., and Petit, C. (2002) Myosin VIIa, Harmonin and Cadherin 23, Three Usher I Gene Products that Cooperate to Shape the Sensory Hair Cell Bundle. EMBO J. 21, 6689-6699. 19. Adato, A., Michel, V., Kikkawa, Y., Reiners, J., Alagramam, K. N., Weil, D., Yonekawa, H., Wolfrum, U., El-Amraoui, A., and Petit, C. (2005) Interactions in the Network of Usher Syndrome Type 1 Proteins. Hum Mol Genet. 14, 347-356. 20. Kros, C. J., Marcotti, W., van Netten, S. M., Self, T. J., Libby, R. T., Brown, S. D., Richardson, G. P., and Steel, K. P. (2002) Reduced Climbing and Increased Slipping Adaptation in Cochlear Hair Cells of Mice with Myo7a Mutations. Nat Neurosci. 5, 41-47. 21. Schwander, M., Xiong, W., Tokita, J., Lelli, A., Elledge, H. M., Kazmierczak, P., Sczaniecka, A., Kolatkar, A., Wiltshire, T., Kuhn, P., Holt, J. R., Kachar, B., Tarantino, L., and Muller, U. (2009) A Mouse Model for Nonsyndromic Deafness (DFNB12) Links Hearing Loss to Defects in Tip Links of Mechanosensory Hair Cells. Proc Natl Acad Sci U S A. 106, 5252-5257. 22. Siemens, J., Lillo, C., Dumont, R. A., Reynolds, A., Williams, D. S., Gillespie, P. G., and Muller, U. (2004) Cadherin 23 is a Component of the Tip Link in Hair-Cell Stereocilia. Nature. 428, 950-955. 23. Caberlotto, E., Michel, V., Foucher, I., Bahloul, A., Goodyear, R. J., Pepermans, E., Michalski, N., Perfettini, I., Alegria-Prevot, O., Chardenoux, S., Do Cruzeiro, M., Hardelin, J. P., Richardson, G. P., Avan, P., Weil, D., and Petit, C. (2011) Usher Type 1G Protein Sans is a Critical Component of the Tip-Link Complex, a Structure Controlling Actin Polymerization in Stereocilia. Proc Natl Acad Sci U S A. 108, 5825-5830. 25. Sengupta, S., George, M., Miller, K. K., Naik, K., Chou, J., Cheatham, M. A., Dallos, P., Naramura, M., Band, H., and Zheng, J. (2009) EHD4 and CDH23 are Interacting Partners in Cochlear Hair Cells. J Biol Chem. 284, 20121-20129. 26. Lagziel, A., Ahmed, Z. M., Schultz, J. M., Morell, R. J., Belyantseva, I. A., and Friedman, T. B. (2005) Spatiotemporal Pattern and Isoforms of Cadherin 23 in Wild Type and Waltzer Mice during Inner Ear Hair Cell Development. Dev Biol. 280, 295-306. 27. Wilson, S. M., Householder, D. B., Coppola, V., Tessarollo, L., Fritzsch, B., Lee, E. C., Goss, D., Carlson, G. A., Copeland, N. G., and Jenkins, N. A. (2001) Mutations in Cdh23 Cause Nonsyndromic Hearing Loss in Waltzer Mice. Genomics. 74, 228-233. 29. Reiners, J., Reidel, B., El-Amraoui, A., Boeda, B., Huber, I., Petit, C., and Wolfrum, U. (2003) Differential Distribution of Harmonin Isoforms and their Possible Role in Usher-1 Protein Complexes in Mammalian Photoreceptor Cells. Invest Ophthalmol Vis Sci. 44, 5006-5015. 30. Reiners, J., Nagel-Wolfrum, K., Jurgens, K., Marker, T., and Wolfrum, U. (2006) Molecular Basis of Human Usher Syndrome: Deciphering the Meshes of the Usher Protein Network Provides Insights into the Pathomechanisms of the Usher Disease. Exp Eye Res. 83, 97-119. 32. Tepass, U., Truong, K., Godt, D., Ikura, M., and Peifer, M. (2000) Cadherins in Embryonic and Neural Morphogenesis. Nat Rev Mol Cell Biol. 1, 91-100. 34. Bahloul, A., Michel, V., Hardelin, J. P., Nouaille, S., Hoos, S., Houdusse, A., England, P., and Petit, C. (2010) Cadherin-23, Myosin VIIa and Harmonin, Encoded by Usher Syndrome Type I Genes, Form a Ternary Complex and Interact with Membrane Phospholipids. Hum Mol Genet. 19, 3557-3565. 36. Kachar, B., Parakkal, M., Kurc, M., Zhao, Y., and Gillespie, P. G. (2000) High-Resolution Structure of Hair-Cell Tip Links. Proc Natl Acad Sci U S A. 97, 13336-13341. 38. Manji, S. S., Miller, K. A., Williams, L. H., Andreasen, L., Siboe, M., Rose, E., Bahlo, M., Kuiper, M., and Dahl, H. H. (2011) An ENU-Induced Mutation of Cdh23 Causes Congenital Hearing Loss, but no Vestibular Dysfunction, in Mice. Am J Pathol. 179, 903-914. 39. Reiners, J., Marker, T., Jurgens, K., Reidel, B., and Wolfrum, U. (2005) Photoreceptor Expression of the Usher Syndrome Type 1 Protein Protocadherin 15 (USH1F) and its Interaction with the Scaffold Protein Harmonin (USH1C). Mol Vis. 11, 347-355. 40. Wagatsuma, M., Kitoh, R., Suzuki, H., Fukuoka, H., Takumi, Y., and Usami, S. (2007) Distribution and Frequencies of CDH23 Mutations in Japanese Patients with Non-Syndromic Hearing Loss. Clin Genet. 72, 339-344. 41. Zheng, Q. Y., Yan, D., Ouyang, X. M., Du, L. L., Yu, H., Chang, B., Johnson, K. R., and Liu, X. Z. (2005) Digenic Inheritance of Deafness Caused by Mutations in Genes Encoding Cadherin 23 and Protocadherin 15 in Mice and Humans. Hum Mol Genet. 14, 103-111. 43. Yonezawa, S., Yoshizaki, N., Kageyama, T., Takahashi, T., Sano, M., Tokita, Y., Masaki, S., Inaguma, Y., Hanai, A., Sakurai, N., Yoshiki, A., Kusakabe, M., Moriyama, A., and Nakayama, A. (2006) Fates of Cdh23/CDH23 with Mutations Affecting the Cytoplasmic Region. Hum Mutat. 27, 88-97. 44. Wada, T., Wakabayashi, Y., Takahashi, S., Ushiki, T., Kikkawa, Y., Yonekawa, H., and Kominami, R. (2001) A Point Mutation in a Cadherin Gene, Cdh23, Causes Deafness in a Novel Mutant, Waltzer Mouse Niigata. Biochem Biophys Res Commun. 283, 113-117. 45. Han, F., Yu, H., Tian, C., Chen, H. E., Benedict-Alderfer, C., Zheng, Y., Wang, Q., Han, X., and Zheng, Q. Y. (2012) A New Mouse Mutant of the Cdh23 Gene with Early-Onset Hearing Loss Facilitates Evaluation of Otoprotection Drugs. Pharmacogenomics J. 12, 30-44. |
|---|
| Science Writers | Anne Murray |
|---|
| Illustrators | Peter Jurek |
|---|
| Authors | Adam Dismang, Tiana Purrington, and Bruce Beutler |
|---|
 Phenotypic Mutation
Phenotypic Mutation